

Important Safety Information
WARNING:
ESAs INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS and TUMOR PROGRESSION OR RECURRENCE
CHRONIC KIDNEY DISEASE:
- In controlled trials, patients experienced greater risks for death, serious adverse cardiovascular reactions, and stroke when administered erythropoiesis-stimulating agents (ESAs) to target a hemoglobin level of greater than 11 g/dL.
- No trial has identified a hemoglobin target level, ESA dose, or dosing strategy that does not increase these risks.
- Use the lowest MIRCERA® dose sufficient to reduce the need for red blood cell (RBC) transfusions.
CANCER:
- MIRCERA® is not indicated and is not recommended for the treatment of anemia due to cancer chemotherapy. A dose-ranging study of MIRCERA® was terminated early because of more deaths among patients receiving MIRCERA® than another ESA.
- ESAs shortened overall survival and/or increased the risk of tumor progression or recurrence in clinical studies in patients with breast, non-small cell lung, head and neck, lymphoid, and cervical cancers.
CONTRAINDICATIONS
MIRCERA® is contraindicated in patients with:
- Uncontrolled hypertension
- Pure red cell aplasia (PRCA) that begins after treatment with MIRCERA® or other erythropoietin protein drugs
- History of serious or severe allergic reactions to MIRCERA® (e.g., anaphylactic reactions, angioedema, bronchospasm, pruritus, skin rash, and urticaria).
INCREASED MORTALITY, MYOCARDIAL INFARCTION, STROKE, AND THROMBOEMBOLISM
- In controlled clinical trials of patients with CKD comparing higher hemoglobin targets (13 to 14 g/dL) to lower targets (9 to 11.3 g/dL), ESAs increased the risk of death, myocardial infarction, stroke, congestive heart failure, thrombosis of hemodialysis vascular access, and other thromboembolic events in the higher target groups.
- Using ESAs to target a hemoglobin level of greater than 11 g/dL increases the risk of serious adverse cardiovascular reactions and has not been shown to provide additional benefit. Use caution in patients with coexistent cardiovascular disease and stroke. Patients with CKD and an insufficient hemoglobin response to ESA therapy may be at even greater risk for cardiovascular reactions and mortality than other patients. A rate of hemoglobin rise of greater than 1 g/dL over 2 weeks may contribute to these risks.
- In controlled clinical trials of patients with cancer, ESAs increased the risks for death and serious adverse cardiovascular reactions. These adverse reactions included myocardial infarction and stroke.
- In controlled clinical trials, ESAs increased the risk of death in patients undergoing coronary artery bypass graft surgery (CABG) and the risk of deep venous thrombosis (DVT) in patients undergoing orthopedic procedures.
INCREASED MORTALITY AND/OR INCREASED RISK OF TUMOR PROGRESSION OR RECURRENCE IN PATIENTS WITH CANCER
- MIRCERA® is not indicated and is not recommended for use in the treatment of anemia due to cancer chemotherapy. A dose-ranging trial of MIRCERA® in 153 patients who were undergoing chemotherapy for non-small cell lung cancer was terminated prematurely because more deaths occurred among patients receiving MIRCERA® than another ESA.
- ESAs resulted in decreased locoregional control/progression-free survival and/or overall survival. These findings were observed in studies of patients with advanced head and neck cancer receiving radiation therapy, in patients receiving chemotherapy for metastatic breast cancer or lymphoid malignancy, and in patients with non-small cell lung cancer or various malignancies who were not receiving chemotherapy or radiotherapy.
HYPERTENSION
- MIRCERA® is contraindicated in patients with uncontrolled hypertension.
- In MIRCERA® clinical studies, approximately 27% of patients with CKD, including patients on dialysis and patients not on dialysis, required intensification of antihypertensive therapy. Hypertensive encephalopathy and/or seizures have been observed in patients with CKD treated with MIRCERA®.
- Appropriately control hypertension prior to initiation of and during treatment with MIRCERA®. Reduce or withhold MIRCERA® if blood pressure becomes difficult to control. Advise patients of the importance of compliance with antihypertensive therapy and dietary restrictions.
SEIZURES
- Seizures have occurred in patients participating in MIRCERA® clinical studies. During the first several months following initiation of MIRCERA®, monitor patients closely for premonitory neurologic symptoms. Advise patients to contact their healthcare practitioner for new-onset seizures, premonitory symptoms, or change in seizure frequency.
LACK OR LOSS OF HEMOGLOBIN RESPONSE TO MIRCERA®
- For lack or loss of hemoglobin response to MIRCERA®, initiate a search for causative factors (e.g., iron deficiency, infection, inflammation, bleeding).
- If typical causes of lack or loss of hemoglobin response are excluded, evaluate for PRCA. In the absence of PRCA, follow dosing recommendations for management of patients with an insufficient response to MIRCERA® therapy.
PURE RED CELL APLASIA
- Cases of PRCA and of severe anemia, with or without other cytopenias that arise following the development of neutralizing antibodies to erythropoietin have been reported in the postmarketing setting in patients treated with MIRCERA®. This has been reported predominantly in patients with CKD receiving ESAs by subcutaneous administration. PRCA was not observed in clinical studies of MIRCERA®.
- PRCA has also been reported in patients receiving ESAs for anemia related to hepatitis C treatment (an indication for which MIRCERA® is not approved).
- If severe anemia and low reticulocyte count develop during treatment with MIRCERA®, withhold MIRCERA® and evaluate patients for neutralizing antibodies to erythropoietin. Serum samples should be obtained at least a month after the last MIRCERA® administration to prevent interference of MIRCERA® with the assay. Contact CSL Vifor at 1-800-576-8295 to perform assays for binding and neutralizing antibodies. Permanently discontinue MIRCERA® in patients who develop PRCA following treatment with MIRCERA® or other erythropoietin protein drugs. Do not switch patients to other ESAs as antibodies may cross-react.
SERIOUS ALLERGIC REACTIONS
- Serious allergic reactions, including anaphylactic reactions, angioedema, bronchospasm, tachycardia, pruritus, skin rash and urticaria have been reported in patients treated with MIRCERA®. If a serious allergic or anaphylactic reaction occurs due to MIRCERA®, immediately and permanently discontinue MIRCERA® and administer appropriate therapy.
SEVERE CUTANEOUS REACTIONS
- Blistering and skin exfoliation reactions including Erythema multiforme and Stevens-Johnson Syndrome (SJS)/Toxic Epidermal Necrolysis (TEN), have been reported in patients treated with ESAs (including MIRCERA®) in the postmarketing setting. Discontinue MIRCERA® therapy immediately if a severe cutaneous reaction, such as SJS/TEN, is suspected.
DIALYSIS MANAGEMENT
- Patients may require adjustments in their dialysis prescription after initiation of MIRCERA®. Patients receiving MIRCERA® may require increased anticoagulation with heparin to prevent clotting of the extracorporeal circuit during hemodialysis.
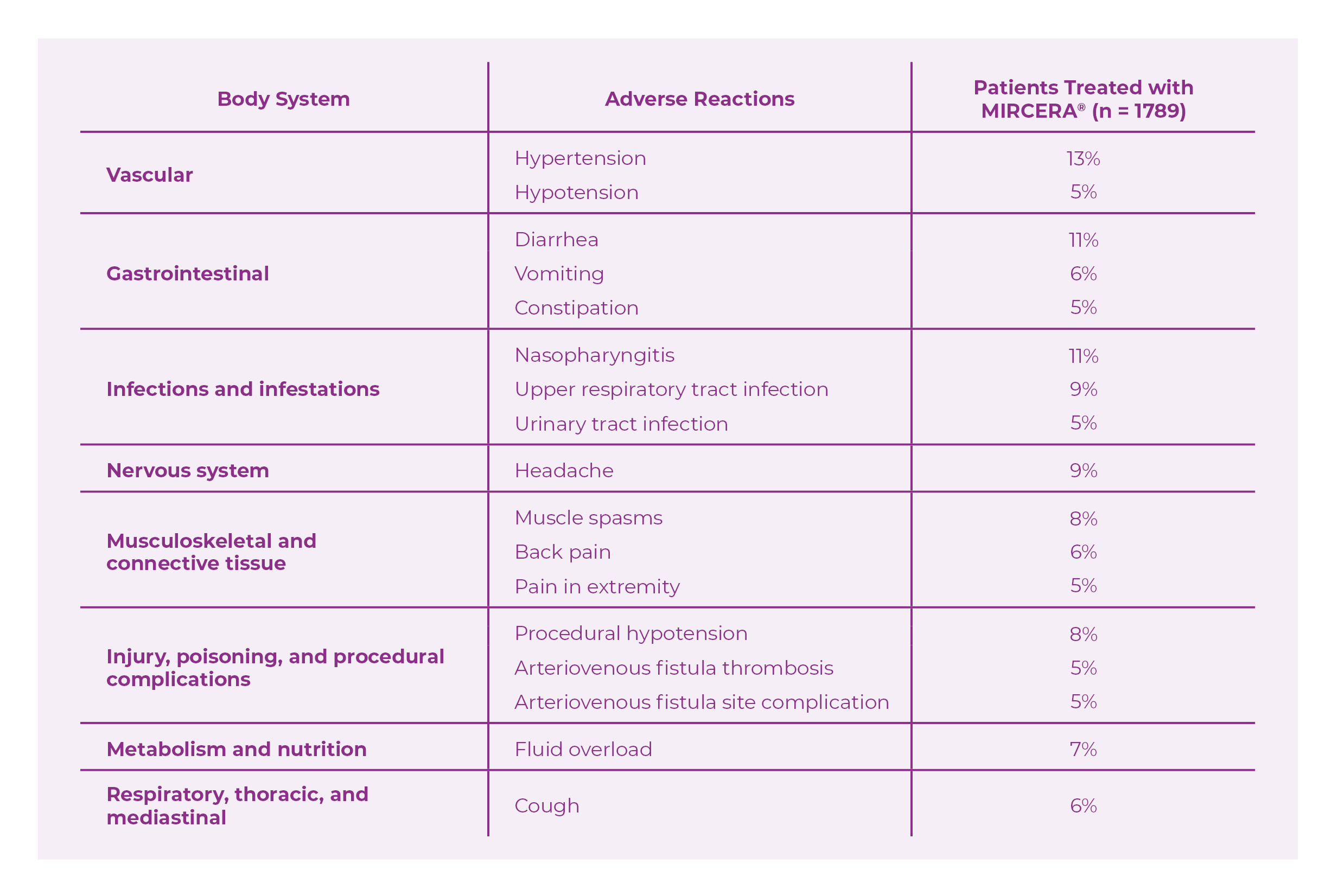
ADVERSE EVENTS IN PATIENTS WITH CHRONIC KIDNEY DISEASE
- Most frequent adverse reactions (≥ 5%) in adult patients with CKD treated with MIRCERA® were hypertension, diarrhea, nasopharyngitis, upper respiratory tract infection, headache, muscle spasms, procedural hypotension, fluid overload, vomiting, back pain, cough, hypotension, constipation, urinary tract infection, pain in extremity, arteriovenous fistula thrombosis, arteriovenous fistula site complication.
- In pediatric patients on hemodialysis, all reported adverse reactions regardless of causality (more than 5% incidence) were headache, nasopharyngitis, hypertension, vomiting, bronchitis, abdominal pain, arteriovenous fistula thrombosis, cough, device related infection, hyperkalemia, pharyngitis, pyrexia, thrombocytopenia, and thrombosis in device.
INDICATIONS AND LIMITATIONS OF USE
- MIRCERA® is indicated for the treatment of anemia associated with chronic kidney disease (CKD) in adult patients on dialysis and adult patients not on dialysis, and pediatric patients 3 months to 17 years of age on dialysis or not on dialysis who are converting from another ESA after their hemoglobin level was stabilized with an ESA.
- MIRCERA® is not indicated and is not recommended for use in the treatment of anemia due to cancer chemotherapy, or as a substitute for RBC transfusions in patients who require immediate correction of anemia.
- MIRCERA® has not been shown to improve quality of life, fatigue, or patient well-being.
Please see full Prescribing Information including Boxed WARNING, and Medication Guide (English, Español) for MIRCERA® (methoxy polyethylene glycol-epoetin beta) Injection, for Intravenous or Subcutaneous Use.
